
Beta-lapachone, a napthoquinone compound originally extracted from the bark of the lapacho tree, has been primarily studied as a possible anti-cancer agent. However, other lines of research have recently revealed that beta-lapachone might be a potent anti-aging and life extension agent as well.
Calorie restriction
Extending healthy lifespan has long been the holy grail of medical research. Decades ago, researchers discovered that restricting calorie intake while providing sufficient nutrients was a very reliable method of extending the healthy lifespan of a wide variety of laboratory animals (1). Life-long calorie restriction can significantly extend the healthy lifespan of yeast, fish, rats, mice, and dogs, as long as all basic nutritional needs are met. Caloric restriction seems to slow down the aging process, extending the period of youthful health. Similar results have been shown in primates. A study of monkeys fed 35% fewer calories since reaching young adulthood reported that caloric restriction significantly extended the healthy lifespan of the monkeys relative to the control group (2).
Although it is easy to feed a captive population a highly nutritious, calorie-restricted diet, convincing free-living animals and humans to comply life-long with such a diet is practically impossible. Therefore, scientists have been studying the mechanism whereby calorie restriction extends life, in hopes of identifying ways to extend life, or at least to extend healthy life, in a more practical way.
Beta-lapachone delays aging
One very interesting study reported that administering beta-lapachone to aging mice prevented age-related declines in motor and cognitive function (3). Although the treated mice did not decrease their food intake or increase their activity, they lost significant amounts of body fat relative to control mice. Interestingly, this same study included a group of calorie-restricted mice as a control. The beta-lapachone-treated mice and the calorie-restricted mice had similar youthful levels of body fat and similar delays in age-related decline. However, in some ways the beta-lapachone treatment improved the health of the mice more than calorie restriction. For example, the blood glucose levels and insulin levels were better in the beta-lapachone-treated mice. Even more interesting was that the beta-lapachone group lived significantly longer than the calorie-restricted group, which lived significantly longer than the control group.
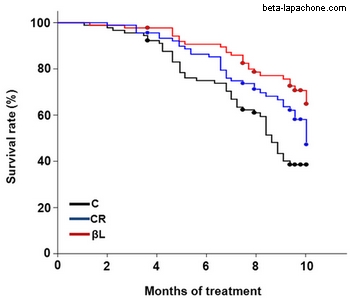
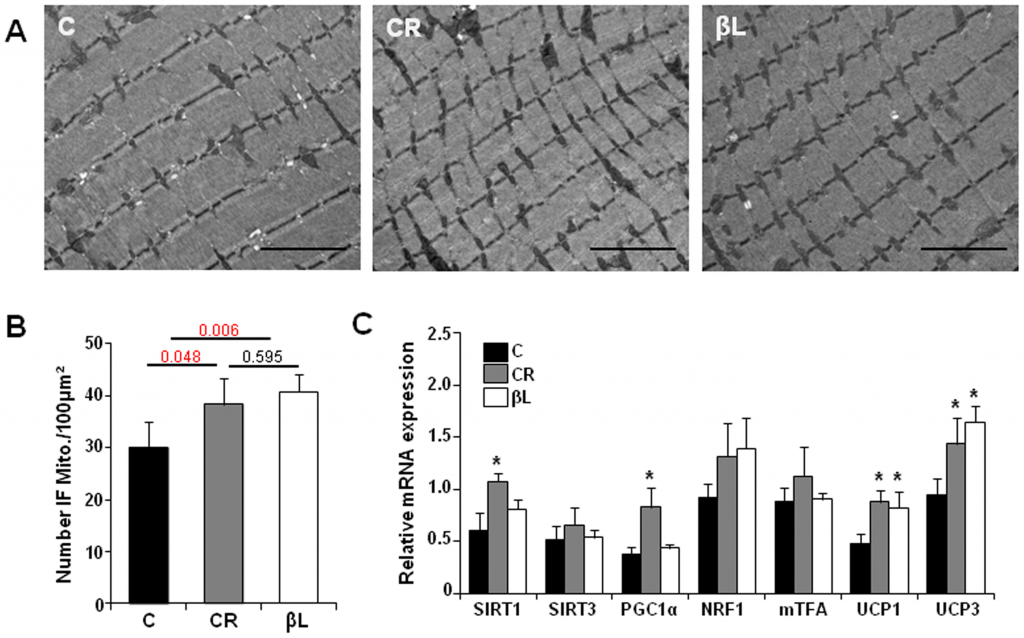
Other interesting point is that beta-lapachone seems to protects the mitochondria structure even better than calorie restriction (fig. B):
NQO1
The enzyme NADH-quinone oxidoreductase 1 (NQO1) has been implicated as a major player in the aging process. NQO1 uses NADH as an electron donor to reduce various substrates, thereby altering the ratio of NAD+/NADH in the cell. Overexpression of NQO1 in yeast increases the lifespan of the yeast (4). Due to technical difficulties in manipulating NQO1 expression in mammals, these types of experiments have not been directly replicated in animal models. However, it has been established that the expression of NQO1 declines with age, and that caloric restriction up-regulates the expression of NQO1 (5).
Beta-lapachone and NAD+
Beta-lapachone has long been known to facilitate the oxidation of NADH to NAD+ by NQO1. During this reaction, the quinone structure of beta-lapachone is able to absorb two electrons from NADH, improving the efficiency of the reaction and altering the ratio of NAD+/NADH in tissues (3).
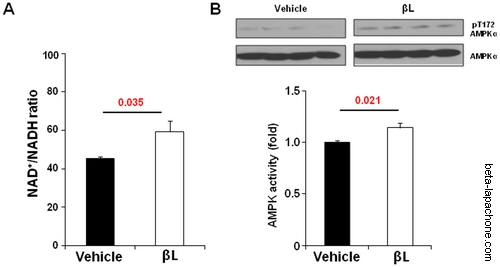
Beta-lapachone increases the NAD+/NADH ratio and activates the AMPK signalling pathways in aged mice : (A) βL increases NAD+/NADH ratio by acute injection. NAD+/NADH ratio was calculated in the tissue of mice intravenously with vehicle (n = 5) or 5 mg/kg βL (n = 5) (B) Effect of βL on phosphorylation of AMPK for EDL (n = 5 per each group). Values represent mean± SEM. doi:10.1371/journal.pone.0047122.g001
High levels of NAD+ relative to NAHD are essential for many processes, such as DNA repair. Activation of SIRT1, a master regulator of aging, induces many processes that are dependent on an abundant supply of NAD+ (6).
Commenting on the study (3) and the link between NAD+ and caloric restriction, Dr. Ross Grant, the author of the study Age Related Changes in NAD+ Metabolism Oxidative Stress and Sirt1 Activity in Wistar Rats, stated :
“The paper provides evidence that supplementation with beta-lapachone increases the oxidation of NADH to NAD+. This mimics calorie restriction (where less NADH is made). This is a good thing as the higher the available NAD+ the better will be the sirtuin associated epigenetic control. However, if the diet is still calorie dense there is still the potential for increased oxidative damage outside of the mitochondria (e.g. NADPH oxidase, immune up-regulation etc) and therefore reductions in NAD+ availability. But beta-lapachone seems to have some potential to positively modulate NAD+”
Beta-lapachone’s ability to directly alter the NAD+/NADH ratio was also shown recently in a landmark experiment in mice. Cisplatin is an important chemotherapeutic agent for treating cancer, but its use is limited by its toxic effect on the kidneys. Beta-lapachone was able to protect the kidneys of mice treated with cisplatin (14). The renal tissues of the treated mice had markedly elevated levels of NAD+, along with reduced levels of inflammatory cytokines and markers of biochemical damage. In a commentary on this study, Lu suggested that the increased levels of NAD+ activate SIRT1, which then activates the same biochemical pathways activated during caloric restriction (15). Calorie restriction is also able to protect the kidneys from cisplatin-induced damage.
Energy dysregulation
During calorie restriction, a low supply of glucose leads to an increase in NAD+ by altering metabolic pathways to produce ATP by oxidizing NADH to NAD+. Beta-lapachone increases the supply of NAD+ relative to NADH by directly acting on NQO1. The improved supply of NAD+ seems to act as a signalling molecule that affects the metabolism of glucose and lipids, restoring a more youthful metabolism to cells and tissues. Dysregulation of energy metabolism is thought to underlie many of the changes associated with aging (7).
Breakdown of nucleus-mitochondrial communication
During aging, the mitochondria begin to malfunction. The mitochondria are cellular organelles responsible for the production of energy in the cell. Studies investigating the mechanism behind the age-related malfunction have identified a gradual loss of mitochondria-specific oxidative phosphorylation subunits. The loss of these subunits seems to be due to a breakdown in communication between the nucleus of the cell and the mitochrondria caused by pseudohypoxia due to the age-related decline in NAD+ (8). This mitchondrial dysfunction leads to changes in metabolism sometimes referred to as ‘Warburg-like reprogramming.’
The Warburg effect
Warburg reprogramming of the metabolism was first discovered in cancer cells. As they progress towards malignancy, their metabolisms change from the normal aerobic glycolysis pathways to pathways in which glucose is fermented into lactate instead. These changes are due to abnormalities in mitochondrial function caused by changes in the growth factor signalling systems of cancerous cells. In cells that have undergone this transition, most of the NAD in the cell is in the NADH form. Originally, it was thought that cancer cells underwent Warburg reprogramming in response to oxygen starvation, but even cancer cells that have plenty of oxygen undergo this metabolic change.
Inefficient metabolism
In normal cells, glucose is transformed into ATP through oxidative phosphorylation in the mitochondria. This is a very efficient energy conversion pathway that relies on a series of electron donations from NADH and FADH2, ultimately transferring the electrons to oxygen. This pathway produces 36 ATP molecules per glucose molecule consumed. Cells that have undergone Warburg reprogramming can only produce 2 ATP molecules per glucose molecule consumed. This metabolic conversion seems to be beneficial to the rapid proliferation of cancer cells. It doesn’t seem to be as beneficial to aging cells.
Reverse the Warburg effect and turn back the clock
In aging mice, restoring a high ratio of NAD+ to NADH can reverse the mitochondrial dysfunction that leads to the Warburg effect in a SIRTI-dependent fashion (8). The initial studies on turning back the aging clock in mice by up-regulating NAD+ relative to NADH were conducted by David Sinclair, an expert on aging who does research at the University of New South Wales and Harvard. In their most recent study, they took old mice and artificially boosted their NAD+ levels by injecting a pharmaceutical agent. The mice were able to transition from the equivalent of a 60-year-old human to a 20-year-old human in about a week on many different physiological measures (8). The NAD+/NADH ratio is clearly what matters in aging.
Cell death
Beta-lapachone has been investigated as an anti-cancer drug. When applied to cancer cells, it induces apoptosis (programmed cell death). This mechanism of action seems to be mediated via the interaction of beta-lapachone and NQO1 on the abnormal mitochondria of cancer cells (10). NQO1 transfers two electrons to beta-lapachone in the process of converting NADH back to NAD+. After receiving the electrons, beta-lapachone quickly converts back to normal, releasing the electrons as reactive oxygen species. In the abnormal environment of cancer cells, what has been called a ‘perpetual futile redox cycle’ can develop as beta-lapachone and NQO1 interact.
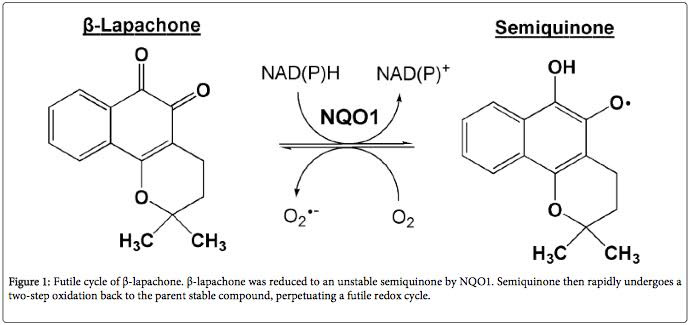
This interaction releases large amounts of reactive oxygen species inside the cancer cells. Therefore, in cells that have dysfunctional mitochondria due to an irreversible condition (e.g., accumulation of mutations such as in cancer cells) beta-lapachone leads to cell death. In cells with a reversible cause of dysfunctional mitochondria, such as aging, beta-lapachone increases the ratio of NAD+ to NADH, reversing the dysfunction and restoring a normal metabolism.
NQO1 regulation pathways
During aging, the ratio of NAD+ to NADH changes in part due to a reduced level of expression of NQO1. Normally, important transcription factors such as nrf1 induce the expression of NQO1. However, in aging tissues, proteins called BET block access to the promoter of NQO1, preventing transcription factors from inducing its expression (11). Lowered levels of NQO1 directly affect the amount of PGC-1a in the cell. PGC-1a levels are regulated by the oxidative state of the cell. When the cell is subjected to oxidative stress, PGC-1a is rapidly oxidized and then degraded by the 20S proteosome system. High levels of NQO1 shift the ratio of NAD+/NADH and protect proteins from being oxidized. PGC-1a plays an important role in regulating mitochondrial function, and higher levels of PGC-1a help prevent age-related mitochondrial dysfunction. Increasing PGC-1a levels through exercise stimulate the tissues to develop a more youthful metabolism, protecting the body against age-related diseases such as obesity, hypertension, and chronic inflammation (12). Declines in NQO1 during aging lead to declines in PGC-1a, strongly contributing to age-related mitochondrial dysfunction. Improving the activity of NQO1 by administering beta-lapachone may reverse this process.
NQO1 and NO
The most common cause of death of aging humans is dysfunction of the cardiovascular system. Chronic inflammation and malfunction of the endothelial cells that line the blood vessels is thought to be a primary cause of cardiovascular disease. Nitric oxide (NO) is used to induce the walls of the blood vessels to relax, improving circulation. Reductions in the amount of NO cause the walls to stiffen and contract, leading to high blood pressure and contributing to the development of cardiovascular disease. In animal models, beta-lapachone is able to reduce blood pressure (13). Beta-lapachone’s ability to improve the activity of NQO1 leads to an increase in endothelial NO synthetase, leading to more NO in the blood vessels. This effect seems to be caused by an NAD+-dependent activation of AMPK, which leads to preservation of GTPCH-1, a rate-limiting enzyme in the synthesis of BH4, a co-factor essential for the function of endothelial NO synthetase. The results of these studies have lead researchers to propose the use of beta-lapachone to treat and possibly prevent high blood pressure and cardiovascular disease in aging humans.
In conclusion, beta-lapachone appears to play many roles in preventing and reversing the signs and symptoms of aging. It mediates these effects by increasing the activity of NQO1, a master regulator of the NAD+/NADH ratio in cells. The NAD+/NADH ratio is what matters. In cells is vitally important for many cellular functions, including regulation of cellular metabolism, DNA repair, apoptosis, and many other aspects of cellular physiology.
References:
(1) https://en.wikipedia.org/wiki/Calorie_restriction
(2) Nat Commun. 2014 Apr 1;5:3557. doi: 10.1038/ncomms4557. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys.
(3) PLoS One. 2012;7(10):e47122. doi: 10.1371/journal.pone.0047122. Epub 2012 Oct 11. Beta-lapachone, a modulator of NAD metabolism, prevents health declines in aged mice.
(4) Aging Cell. 2009 Apr;8(2):140-51. doi: 10.1111/j.1474-9726.2009.00461.x. Epub 2009 Feb 23. NQR1 controls lifespan by regulating the promotion of respiratory metabolism in yeast.
(5) Proc Natl Acad Sci U S A. 2008 Feb 19;105(7):2325-30. doi: 10.1073/pnas.0712162105. Epub 2008 Feb 19. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction.
(6) The Durk Pearson & Sandy Shaw®. Life Extension NewsTM. Volume 16 No. 8 • September 2013. It’s a NAD+, NAD+, NAD+, NAD+ World
(7) FASEB J. 2001 Jan;15(1):108-114. The role of leptin resistance in the lipid abnormalities of aging.
(8) Cell. 2013 Dec 19;155(7):1624-38. doi: 10.1016/j.cell.2013.11.037. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging.
(9) Cancer Cell. 2012 Mar 20; 21(3): 297–308. doi: 10.1016/j.ccr.2012.02.014. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate
(10) Toxicol Appl Pharmacol. 2014 Dec 15;281(3):285-93. doi: 10.1016/j.taap.2014.10.012. Epub 2014 Oct 29. Mechanistic studies of cancer cell mitochondria- and NQO1-mediated redox activation of beta-lapachone, a potentially novel anticancer agent.
(11) J Immunol. 2014 May 15;192(10):4913-20. doi: 10.4049/jimmunol.1301984. Epub 2014 Apr 14. Bromodomain and extraterminal proteins suppress NF-E2-related factor 2-mediated antioxidant gene expression.
(12) Proc Natl Acad Sci U S A. 2009 Dec 1;106(48):20405-10. doi: 10.1073/pnas.0911570106. Epub 2009 Nov 16. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging.
(13) J Hypertens. 2014 Feb;32(2):306-17. doi: 10.1097/HJH.0000000000000018. Enhanced activation of NAD(P)H: quinone oxidoreductase 1 attenuates spontaneous hypertension by improvement of endothelial nitric oxide synthase coupling via tumor suppressor kinase liver kinase B1/adenosine 5′-monophosphate-activated protein kinase-mediated guanosine 5′-triphosphate cyclohydrolase 1 preservation.
(14) Kidney Int. 2014 Mar; 85(3): 547–560. Published online 2013 Sep 11. doi: 10.1038/ki.2013.330 PMCID: PMC3944666 Pharmacological activation of NQO1 increases NAD+ levels and attenuates cisplatin-mediated acute kidney injury in mice
(15) Kidney International (2014) 85, 496–498; doi:10.1038/ki.2013.419 β-Lapachone ameliorates murine cisplatin nephrotoxicity: NAD+, NQO1, and SIRT1 at the crossroads of metabolism, injury, and inflammation
What is the best source and bioavailability of beta lapachone for use as an antiaging supplement ? Where can it be purchased?
I have identified good beta-lapachone source, but first I would like to complete this anti aging topic and also make a new article about beta-lapachone safety and even suggested dosage based on the few studies we hold. Then I will check for a good quality source
Thank!
Hi there, still researching ? Do you have new infos about good beta-lapachone source ? Krisztian
Hello,
we have a source but bit pricey : >98% pure (such the one used in the studies) and 250$ per gram. This is the less expensive we can get and the purest